

Définition du syndrome et prévalence:
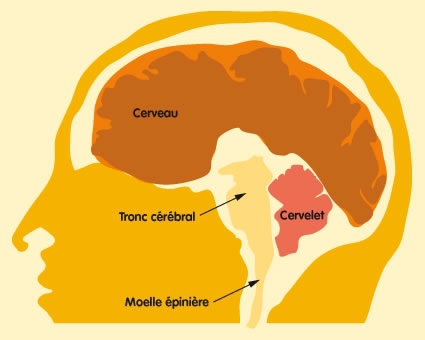
Le syndrome de Joubert, décrit en 1969 par Joubert et Eisenring, est une maladie génétique rare due à une malformation du tronc cérébral et une agénésie (absence de formation) ou une hypoplasie (développement insuffisant) de la partie médiane du cervelet (vermis cérébelleux).
Le tronc cérébral et le cervelet sont deux organes situés à la base du cerveau.
Le syndrome de Joubert touche aussi bien les filles que les garçons et on estime qu’environ une naissance sur 100 000 est concernée.
Voir la définition complète sur le site Orpha.net en cliquant ici
Signes cliniques et évolution:
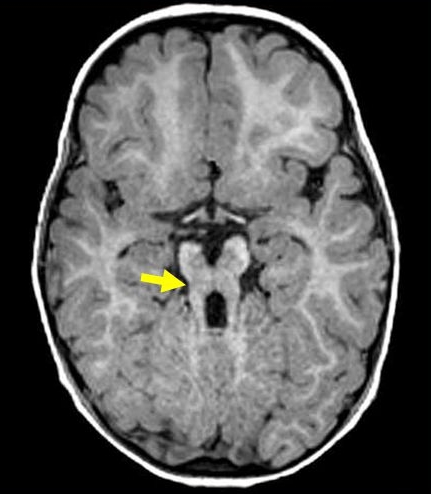
Le diagnostic repose sur les données cliniques et la présence, sur les clichés d'Imagerie par Résonance Magnétique (IRM), du « signe de la dent molaire ».
Au cours de la période néonatale, le syndrome de Joubert se manifeste par une hypotonie (enfant mou), des troubles du rythme respiratoire comme des tachypnées (accélération du rythme respiratoire) et/ou des apnées (pauses de la respiration), et par des mouvements anormaux des yeux (nystagmus).
Durant la petite enfance, un retard du développement moteur est fréquent, entraînant un retard de l’acquisition des stations assise, debout, et de la marche. Une ataxie cérébelleuse (démarche titubante et déséquilibre) apparaît généralement.
Les facultés intellectuelles sont variables, allant d'un déficit intellectuel sévère à une intelligence normale, et les difficultés d’apprentissage sont fréquentes. L'examen neuro-ophtalmologique peut révéler une apraxie oculomotrice (manque de coordination entre les mouvements des yeux et le cerveau).
Les difficultés d'élocution peuvent gêner la parole et nécessiter une prise en charge spécifique.
Les autres manifestations possibles sont des convulsions chez certains patients, une dystrophie rétinienne pouvant mener à la cécité, une dysplasie rénale multikystique ou une néphronophtise (atteinte des reins évoluant vers l'insuffisance rénale), une atteinte hépatique et une polydactylie (doigts surnuméraires).
Les manifestations du syndrome de Joubert sont variables d'un enfant à l'autre et chaque enfant évolue à son rythme.

Mode de transmission et génétique:
Le syndrome de Joubert se transmet dans la plupart des cas sur un mode autosomique récessif, ce qui signifie que le syndrome s’exprime et que l’enfant est atteint lorsque les deux copies du gène présentes chez l’enfant (celle venant du père et celle venant de la mère) ont des mutations ou des altérations.
Les parents d’un enfant atteint du syndrome de Joubert
sont tous les deux porteurs d’une copie altérée du gène et d’une copie non altérée, on dit qu’ils sont hétérozygotes, le syndrome ne s’exprime pas chez eux (ils sont « porteurs
sains »).
A chaque naissance, les parents d’un enfant atteint du syndrome de Joubert ont une probabilité de 25% d’avoir un enfant malade, 50% d’avoir un enfant porteur mais non malade, et de 25% d’avoir un enfant non porteur de la maladie.
Un conseil génétique est préconisé et un prélèvement de villosités choriales (choriocentèse) peut être envisagé en vue d’une future grossesse lorsque le gène mis en cause a été identifié par une étude génétique avec prélèvement sanguin chez les parents et l’enfant.
Il existe également des mutations dites spontanées.

Suivi et prise en charge:
Le suivi des enfants atteints par le syndrome de Joubert est souvent assuré par un pédiatre, un neuropédiatre, ou un généticien.
Sa prise en charge est pluridisciplinaire. Les séances de kinésithérapie spécialisée, d'ergothérapie, d'orthophonie, d'orthoptie, de psychomotricité et de suivi par un éducateur visent à améliorer l'hypotonie, le retard du développement moteur et à améliorer les conditions de vie. Le suivi médical en neuropédiatrie, hépatologie, néphrologie et ophtalmologie peut prévenir des risques et des évolutions des différentes manifestations du syndrome.
Différents centres de référence propres à chaque discipline peuvent être consultés et prendre en charge le suivi des enfants concernés.
Enfin, des suivis et bilans psychologiques et neuropsychologiques peuvent être proposés aux enfants et à leur famille.

La recherche et l'espoir:
A ce jour, une vingtaine de gènes impliqués dans le syndrome de Joubert ont été identifiés.
Cependant, la recherche se poursuit pour recenser de nouveaux gènes responsables du syndrome et mieux cerner leur rôle, leur fonctionnement et leur implication dans cette maladie.
De plus, les progrès de la recherche dans chaque discipline impliquée sont autant d’espoir de soulager les malades.

Mahé et le syndrome :
Mahé est atteint du syndrome de Joubert avec atteinte rénale et oculaire. L’IRM réalisée à ses deux mois a confirmé la présence du signe de la dent molaire. Son hypotonie, son nystagmus et ses kystes rénaux néonataux furent des signes évocateurs de la maladie.
Une étude génétique réalisée après prélèvements sanguins chez Mahé et ses parents a permis d’identifier le gène CEP290/NPHP6 localisé sur le chromosome 12 comme porteur des mutations transmises par ses parents.
Mahé est suivi régulièrement par un pédiatre à Metz, l'Institut d'Education Sensoriel de Metz, un neuropédiatre à Strasbourg, un néphrologue à Nancy, le Centre de référence des Affections rares en Génétique Ophtalmologique de Strasbourg, le Centre de référence des Malformations et Maladies Congénitales du Cervelet de Paris.
Les diverses séances hebdomadaires de kinésithérapie, de rééducation, d'orthophonie et de psychomotricité permettent à Mahé de progresser chaque jour un peu plus et d’impressionner ses parents malgré les difficultés liées à sa cécité.
Liens utiles:
http://anomaliescervelet.aphp.fr/

Association "Mahé de la tête aux pieds" - Siège social: 6, rue du Général Decaen 57070 METZ
Inscrite au Registre des associations du Tribunal d’Instance de Metz le 30 août 2013 – Volume 161, Folio n°79
© 2014 Association "Mahé de la tête aux pieds" - Tous droits réservés